Download this page as a pdf by clicking here.
1. What is erythropoietic protoporphyria?
2. What causes EPP and XLP?
3. What are the symptoms of EPP and XLP?
4. What do EPP and XLP look like?
5. How are EPP and XLP diagnosed?
6. Are EPP and XLP inherited?
7. Are EPP and XLP dangerous diseases?
8. Protection from light in EPP and XLP
9. How can EPP and XLP be treated?
10. Supportive therapies and interventions in EPP and XLP
11. Additional information on EPP and XLP
12. Can EPP and XLP be cured?
13. Where can I get more information about EPP and XLP?
1. What is erythropoietic protoporphyria (EPP and XLP)?
EPP is an ultra-rare disease that was first described in 1961. People with EPP suffer from skin pain that starts after they have been outdoors in the sun or exposed to strong artificial light. The symptoms start in early childhood and can be very painful. EPP is an inherited disease. It has been found in one out of 58,000 to 200,000 people in Europe but is rare in people from Africa. The symptoms of EPP is due to a build-up of high levels of the natural product called protoporphyrin during the making of red blood cells in the bone marrow.
2. What causes EPP and XLP?
Patients with EPP have a shortage of an enzyme called ferrochelatase, which adds iron to protoporphyrin to make haem. An enzyme is a protein that helps to convert one chemical into another chemical inside the cell. The shortage of ferrochelatase causes protoporphyrin to build up in the red blood cells.
In XLP the increased protoporphyrin is caused by increased activity of the very first enzyme in the production of haem, ALA synthase 2. This also causes protoporphyrin to build up to in the red blood cells. In very rare cases, another gene may be involved and may have a different inheritance pattern.
As blood passes through the small blood vessels in the skin, protoporphyrin can absorb the energy from light. This sets off a phototoxic chemical reaction that causes tissue damage, itching and burning pain. If the blood vessels are severely damaged, blood fluids leak into the surrounding tissue, causing swelling and reddening of the skin.
The part of sunlight that protoporphyrin absorbs is different from that which causes normal sunburn. Sunburn is caused by invisible ultraviolet (UV) radiation, but in EPP the skin is sensitive to visible light. The light that causes the pain in EPP, unlike the one causing sunburn, can pass through window glass.
3. What are the symptoms of EPP and XLP?
Exposure of the skin to bright light causes the skin first to tingle and itch, and later to become extremely painful, red and swollen. The symptoms usually start within a few minutes of light exposure, but this can vary between individuals and also depend on how strong the light is. Symptoms can take hours or days to go away completely and during this time the skin may be extremely sensitive to light, temperature, touch or wind. The light does not need to be direct: light which is reflected on water, snow and sand, or which passes through window glass can also trigger a phototoxic reaction.
EPP usually starts in childhood. Infants can cry or scream after being taken out into the sunlight. Older children may complain of burning, wave their hands in the air, or put them in cold water to try to relieve the pain. EPP and XLP usually affect males and females equally.
Most individuals with EPP and XLP say that their quality of life is poor because they cannot go outdoors with others, they need to wear gloves, hats and even masks, they have days with severe pain, are anxious and feel poorly understood by outsiders.
4. What do EPP and XLP look like?
Even when the pain is severe, the skin usually looks normal. Sometimes there can be swelling of the skin, initially with the appearance of a nettle rash and some redness. With time, the skin over the knuckles may become thick, and small scars may be found on the nose, cheeks and the back of the hands.
5. How are EPP and XLP diagnosed?
A diagnosis of EPP or XLP should be suspected when a person complains of pain in light exposed skin. For the diagnosis, a blood sample should be collected from the patient, protected from light and sent to a laboratory specialised in porphyria diagnostics. The blood test measures the amount of protoporphyrin in the red blood cells (erythrocyte protoporphyrin). A urine porphyrin test is of no use when testing for EPP or XLP. For confirmation of the diagnosis or family counseling, genetic testing is sometimes performed.
Since EPP and XLP are such rare diseases, most doctors are not familiar with them. It may take years before EPP is suspected and a diagnosis is made.
6. Are EPP and XLP inherited?
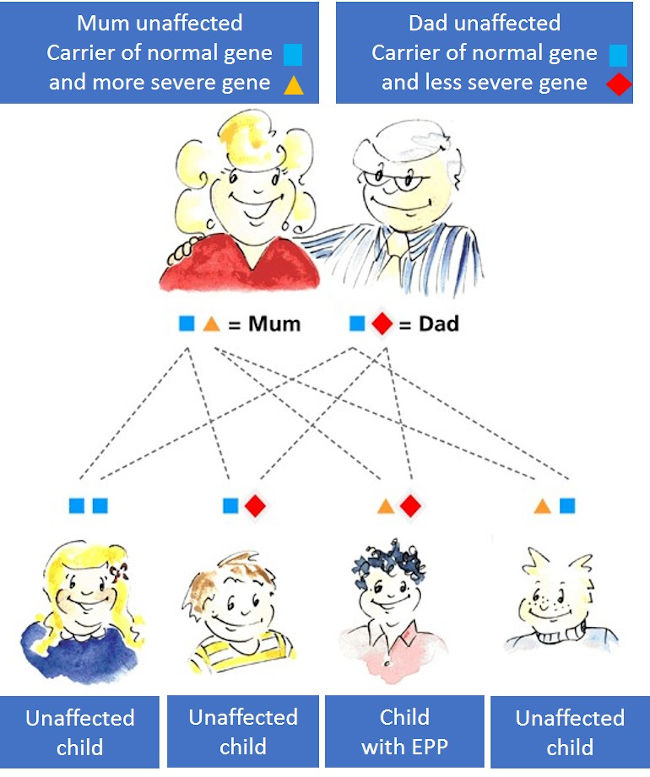
In most cases EPP is inherited, but many EPP patients do not know of other members of their family with the disease. This is because we all have two genes for ferrochelatase, one inherited from our mother and one from our father, and both genes have to be faulty to develop EPP. Most EPP patients inherit a severely affected gene from one parent and a less severely affected gene from the other parent. One abnormal gene alone is not sufficient to cause EPP. The more severely affected gene is very uncommon, but the less severe gene is present in about 10% of people in European populations. The more severe gene may be inherited down a family line but EPP does not occur until it “meets” the common but less severe gene again. This is why EPP sometimes skips generations before another family member develops EPP.
If you have EPP, the risk that your children will develop EPP depends on the genes your partner carries, but is usually small. If your partner belongs to the nine out of ten people who have normal genes, there is virtually no risk. If your partner carries the less severe gene (a 1 in 10 chance), the risk of a child inheriting EPP is one in four. We recommend you ask a clinical genetics service or a porphyria specialist for advice on inheritance and genetic testing.
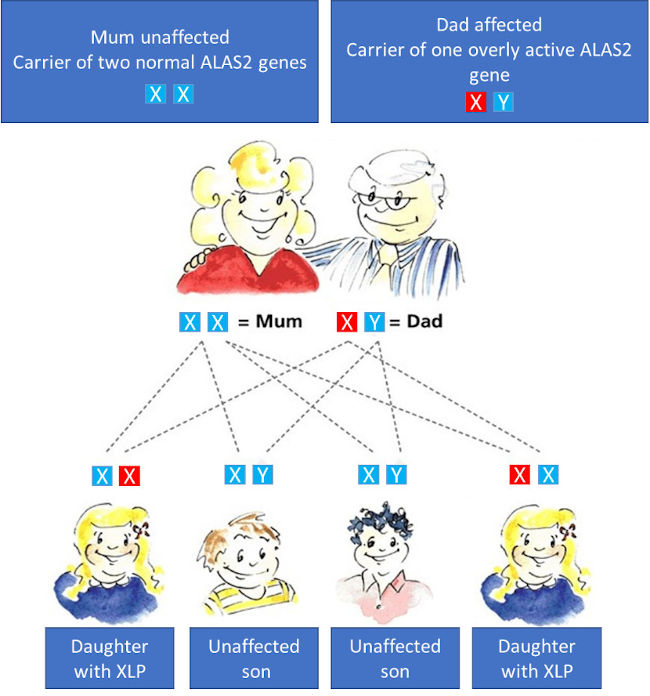
The gene for ALAS2, which is overactive in XLP, is located on the X-chromosome. This X-chromosome together with a second X or a Y chromosome decides the sex of a child (XX chromosomes are female, XY chromosomes are male). In the case of XLP, one overly active gene is sufficient to cause the disease. Fathers with XLP will pass on their condition to all their daughters, but cannot pass it onto a son (who inherits his fathers unaffected Y-chromosome). (see below)
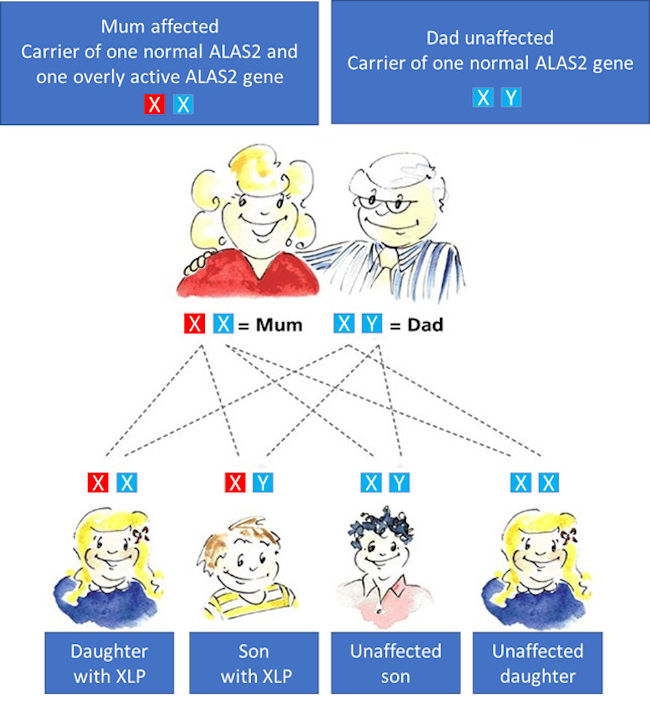
Mothers affected by XLP will pass on their overly active ALAS2 gene to half of their daughters and sons. On average 50 % of offspring from an affected parent are affected by XLP. (see below)
Very rarely, EPP or XLP can occur later in life, often associated with an underlying blood malignancy.
7. Are EPP and XLP dangerous diseases?
A small number of EPP and XLP patients may develop liver damage. It is not possible to foresee who will be affected but fortunately this is rare. If you become more sensitive to light, become tired or the whites of your eyes start to turn yellow, you should ask your doctor to test your liver function as soon as possible. EPP and XLP patients also get gall stones more commonly than the general population.
Pregnancy and child-bearing is not associated with any additional risks and the expected outcome for the baby is normal. Sometimes women with EPP can stay out longer in sunlight while they are pregnant.
Because EPP patients avoid sunlight, they often have too little vitamin D, and their bones therefore contain less calcium than normal.
EPP patients are commonly slightly anaemic, with low blood haemoglobin and iron levels. However, taking iron supplements can increase sensitivity to light, and iron supplements should only be taken when prescribed by your doctor (see below).
You should visit your doctor for regular check-ups – at least once a year. Your doctor may monitor your blood protoporphyrin, your vitamin D level, the way your liver is working, and other organ functions by yearly blood tests. The purpose of regular monitoring is to detect any abnormalities before they develop into risky conditions.
8. Protection from light in EPP and XLP
From childhood onwards, patients with EPP try to avoid unnecessary exposure to sunlight and strong artificial light sources. Protective clothing such as hats, long sleeves, gloves and trousers are beneficial. Dark and closely woven fabrics protect better than thin clothes with light colours. Sunscreens mainly protect against UV radiation and do not protect against light that causes EPP symptoms. Windows in houses and cars offer no protection since harmful visible light passes through most windows. Teachers need to be informed about the diagnosis and the need for special requirements such as a work place away from windows in schools and the right to retreat from outdoor activities.
9. How can EPP and XLP be treated?
Afamelanotide (Scenesse®) is currently the only treatment for which safety and effectiveness have been demonstrated in clinical trials. It was authorised for use in the European Union in 2014 and in the USA in 2019. Treatment with afamelanotide significantly increases the time EPP patients can spend in direct sunlight without pain and increases their quality of life. Afamelanotide is administered as an implant which is injected under the skin above the hip every two months. Mild side effects have been reported. Afamelanotide is not yet available in all European countries.
Substances and interventions with currently unproven effectiveness
A variety of interventions and substances have been used to improve the symptoms of EPP and XLP, although safety and effectiveness have not been studied through clinical trials. The most commonly used substances and interventions are discussed below.
- Beta-carotene
Beta-carotene used to be prescribed for EPP but few people with EPP found taking it helpful. Beta-carotene is derived from the chemical that makes carrots orange and usually give the skin a slightly orange tone. You should discuss the risks and benefits of this option with a porphyria specialist before embarking on treatment. - Antihistamines
Antihistamines, which are mainly used for allergies, may help in case of a nettle rash. Some patients report an additional beneficial effect of hydrocortisone cream on red or swollen skin. Older antihistamines that have drowsiness as a side-effect, may also help sleeping after a mild phototoxic reaction. - Phototherapy
Narrow-band UVB and PUVA therapy are ultraviolet light treatments that are given for different skin conditions in dermatology departments. In EPP they have been used to increase light tolerance by allowing the skin to thicken slightly and develop a tan. Therapy involves careful exposure to increasing doses of controlled UVB radiation, usually three times per week for at least five weeks during the spring. This may be impractical and inconvenient for some patients. Phototherapy should always be directed by a trained photodermaologist. Patients are strongly advised not to use self tanning salons, as these light sources can trigger phototoxic reactions. - Other treatments are also under investigation.
10. Supportive therapies and interventions
- Relief from a painful phototoxic reaction
The severe burning pain in skin after light exposure is difficult to relieve. Many EPP patients report temporary relief by cold baths, cold wet dressings or remedies such as aloe vera. Some patients report considerable relief from the pain by warm water or vapour (steam). No pain medicine is especially effective. The choice and testing out of pain medicines should be done together with your doctor. - Vitamin D
Vitamin D is mainly produced in sun exposed skin, but some also comes from food such as oily fish, eggs, meat and cereals. Individuals who avoid the sun often develop vitamin D deficiency and all EPP patients should take vitamin D at the recommended dose. - Iron supplements
Many patients with EPP have slightly low iron (ferritin) and haemoglobin values. It may therefore be difficult for doctors to figure out if you have a true iron deficiency anaemia and need iron supplementation. Being able to compare with previous blood and iron values helps. Iron deficiency that is causing symptoms due to anaemia should be treated as in any other patient. However in EPP iron supplementation may speed up the production of red blood cells in the bone marrow and increase the protoporphyrin levels in the blood. This may lead to increased sensitivity to light. This does not happen in XLP patients, where iron supplements tend to lead to a reduction in blood protoporphyrin levels. - Sunscreens
As visible light causes the problems in EPP and XLP, widely available sunscreens that protect against ultraviolet light (particularly UVB) are not very effective. - Light filters
Most window filters for cars and offices are designed to block ultraviolet light. Even if they are tinted in smoky or grey colours they let most of the visible light pass through. Filters that selectively block the most harmful light in EPP are yellow. These filters can be applied to windows in homes, schools or offices or even on car side windows. Before applying films to car windows, you should check with traffic authorities to see which films are acceptable according to local traffic laws. - Liver protection
Excess consumption of alcohol or other substances that might damage the liver must be avoided, and you should be vaccinated against hepatitis A and B.
11. Additional information
- Can certain drugs worsen EPP?
EPP differs from the acute hepatic porphyrias that can be made worse by certain medications. Unless allergic to a medication, individuals with EPP can take any form of medication that their health requires. - Laser treatment
Laser treatments for eye surgery or hair removal have not been reported to be a problem in EPP. The blue lasers used in for dental cavity repair can cause burns in the gums. Dentists should be informed of the EPP diagnosis so that they can direct the laser beam with care. - Light sources in health care
Covering operating lights with yellow filters is not necessary for most operations, however, health care staff should always be told that you have a diagnosis of EPP to avoid unnecessarily powerful lighting for longer surgeries. Examination of the inside of the body using a lighted, flexible instrument called an endoscope is also safe.
For operations or endoscopy on patients with severe EPP-related liver disease the doctors should seek advice from a porphyria specialist, as the operating lamps should be covered with yellow filters. Porphyria specialist centres can provide further advice when necessary.12. Can EPP and XLP be cured?
At present there is no cure for EPP other than bone marrow transplantation. This treatment is associated with such high risks of complications and even death that it is only used in very special and complicated situations.
13. Where can I get more information about EPP and XLP?
The information on this website has been written and updated by a group of porphyria specialists working together in Ipnet and checked by representatives of patient organisations. Although several other sources of information can be found on the internet, they may not have been written or examined by porphyria specialists. Patient associations can be a good point of contact for information, networking and support. Porphyria patient groups in different European countries are listed on the International Porphyria Network website and the Global Porphyria Advocacy Coalition (GPAC) website.
Revised May 2021