Laden Sie diese Seite als PDF herunter, indem Sie hier klicken.
Inhalt
1. Was ist erythropoetische Protoporphyrie?
2. Was verursacht EPP und XLP?
3. Was sind die Symptome von EPP und XLP?
4. Wie sehen EPP und XLP aus?
5. Wie werden EPP und XLP diagnostiziert?
6. Werden EPP und XLP vererbt?
7. Sind EPP und XLP gefährliche Krankheiten?
8. Lichtschutz in EPP und XLP
9. Wie können EPP und XLP behandelt werden?
10. Unterstützende Therapien und Interventionen bei EPP und XLP
11. Zusätzliche Informationen zu EPP und XLP
12. Können EPP und XLP geheilt werden?
13. Wo erhalte ich weitere Informationen zu EPP und XLP?
1. Was ist erythropoetische Protoporphyrie (EPP und XLP)?
EPP ist eine extrem seltene Erkrankung, die erstmals 1961 beschrieben wurde. Menschen mit EPP leiden unter Hautschmerzen, die nach einem Aufenthalt draußen in der Sonne oder in starkem künstlichem Licht auftreten. Die Symptome beginnen in der frühen Kindheit und können sehr schmerzhaft sein. EPP ist eine Erbkrankheit, die bei einem von 58.000 bis 200.000 Menschen in Europa auftritt, bei Menschen aus Afrika aber selten ist. Die Symptome von EPP sind auf eine Anhäufung hoher Konzentrationen der natürlichen Verbindung Protoporphyrin während der Bildung roter Blutkörperchen im Knochenmark zurückzuführen.2. Was verursacht EPP und XLP?
Patienten mit EPP haben einen Mangel an einem Enzym namens Ferrochelatase, das Eisen zu Protoporphyrin hinzufügt, um Häm zu bilden. Ein Enzym ist ein Protein, das hilft, in der Zelle eine chemische Verbindung in eine andere Verbindung umzuwandeln. Der Mangel an Ferrochelatase führt dazu, dass sich Protoporphyrin in den roten Blutkörperchen ansammelt.Bei XLP wird das erhöhte Protoporphyrin durch eine erhöhte Aktivität des allerersten Enzyms bei der Bildung von Häm, der ALA-Synthase 2, verursacht. Dies führt auch dazu, dass sich Protoporphyrin in den roten Blutkörperchen ansammelt. In sehr seltenen Fällen kann ein anderes Gen mit anderem Vererbungsmuster beteiligt sein.
Wenn Blut durch die kleinen Blutgefäße in der Haut fließt, kann Protoporphyrin die Energie des Lichts absorbieren. Dies löst eine phototoxische chemische Reaktion aus, die Gewebeschäden, Juckreiz und brennende Schmerzen verursacht. Sind die Blutgefäße stark geschädigt, dringt Blutflüssigkeit in das umliegende Gewebe ein, was zu Schwellungen und Rötungen der Haut führt.
Der Teil des Sonnenlichts, den Protoporphyrin absorbiert, unterscheidet sich von dem, der normalen Sonnenbrand verursacht. Sonnenbrand wird durch unsichtbare ultraviolette (UV) Strahlung verursacht, aber bei EPP reagiert die Haut empfindlich auf sichtbares Licht. Das Licht, das die Schmerzen bei EPP verursacht, kann im Gegensatz zu dem, das Sonnenbrand verursacht, durch Fensterglas dringen.
3. Was sind die Symptome von EPP und XLP?
Wenn die Haut hellem Licht ausgesetzt wird, kribbelt und juckt sie zuerst und wird später extrem schmerzhaft, gerötet und geschwollen. Die Symptome beginnen normalerweise innerhalb weniger Minuten nach der Lichteinwirkung, aber dies kann von Person zu Person variieren und hängt auch davon ab, wie stark das Licht ist. Es kann Stunden oder Tage dauern, bis die Symptome vollständig verschwinden, und während dieser Zeit kann die Haut extrem licht-, temperatur-, berührungs- oder windempfindlich sein. Das Licht muss nicht direkt auf die Haut scheinen: auch Licht, das auf Wasser, Schnee und Sand reflektiert wird oder durch Fensterglas dringt, kann eine phototoxische Reaktion auslösen.EPP beginnt normalerweise in der Kindheit. Säuglinge können weinen oder schreien, nachdem sie Sonnenlicht ausgesetzt wurden. Ältere Kinder klagen möglicherweise über Brennen, wedeln mit den Händen in der Luft oder legen sie in kaltes Wasser, um die Schmerzen zu lindern. EPP und XLP betreffen in der Regel Männer und Frauen gleichermaßen.
Die meisten Menschen mit EPP und XLP geben an, dass ihre Lebensqualität schlecht ist, weil sie nicht mit anderen ins Freie gehen können, Handschuhe, Mützen und sogar Masken tragen müssen, Tage mit starken Schmerzen haben, ängstlich sind und sich von Außenstehenden schlecht verstanden fühlen.
4. Wie sehen EPP und XLP aus?
Selbst bei starken Schmerzen sieht die Haut meist normal aus. Manchmal kann es zu einer Schwellung der Haut kommen, zunächst mit dem Auftreten eines Nesselausschlags und einer Rötung. Mit der Zeit kann die Haut über den Knöcheln dick werden und kleine Narben an Nase, Wangen und Handrücken entstehen.5. Wie werden EPP und XLP diagnostiziert?
Eine Diagnose von EPP oder XLP sollte vermutet werden, wenn eine Person über Schmerzen in lichtexponierter Haut klagt. Zur Diagnose sollte dem Patienten eine Blutprobe entnommen, vor Licht geschützt und an ein auf Porphyriediagnostik spezialisiertes Labor geschickt werden. Der Bluttest misst die Menge an Protoporphyrin in den roten Blutkörperchen (Erythrozyten-Protoporphyrin). Ein Urin-Porphyrin-Test ist beim Testen auf EPP oder XLP nutzlos. Zur Bestätigung der Diagnose oder im Rahmen einer Familienberatung werden manchmal Gentests durchgeführt.Da EPP und XLP so seltene Krankheiten sind, sind die meisten Ärzte damit nicht vertraut. Es kann Jahre dauern, bis ein Verdacht auf EPP besteht und eine Diagnose gestellt wird.
6. Werden EPP und XLP vererbt?
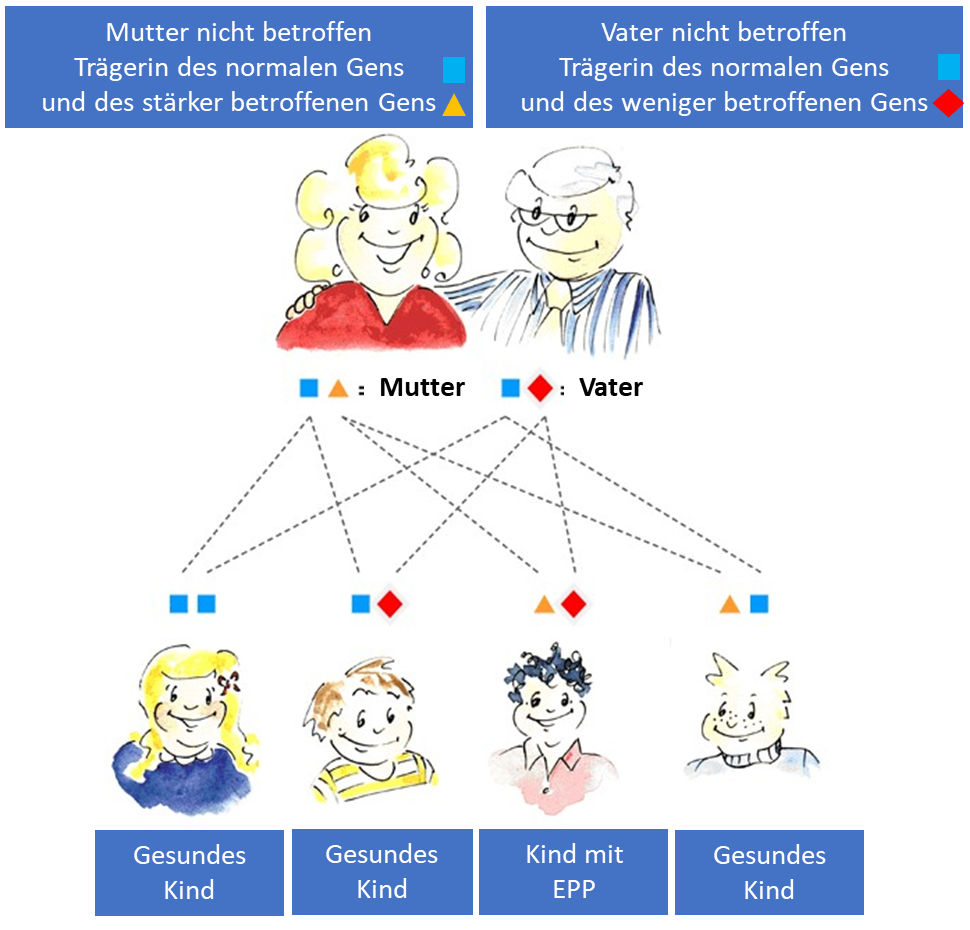
In den meisten Fällen wird EPP vererbt, aber viele EPP-Patienten kennen keine anderen Familienmitglieder mit der Krankheit. Dies liegt daran, dass wir alle zwei Gene für Ferrochelatase haben, eines von unserer Mutter und eines von unserem Vater, und beide Gene müssen fehlerhaft sein, um EPP zu entwickeln. Die meisten EPP-Patienten erben ein stark betroffenes Gen von einem Elternteil und ein weniger stark betroffenes Gen von dem anderen Elternteil. Ein defektes Gen allein reicht nicht aus, um EPP zu verursachen. Das stärker betroffene Gen ist sehr selten, aber das weniger betroffene Gen ist bei etwa 10 % der Menschen in der europäischen Bevölkerung vorhanden. Das stärker betroffenene Gen kann über eine Familienlinie vererbt werden, aber EPP tritt erst auf, wenn es auf das weniger betroffene Gen „trifft“. Aus diesem Grund überspringt EPP manchmal Generationen, bevor ein weiteres Familienmitglied EPP entwickelt.Wenn Sie an EPP leiden, hängt das Risiko, dass Ihre Kinder EPP entwickeln, von den Genen Ihres Partners ab, ist aber normalerweise gering. Gehört Ihr Partner zu den neun von zehn Menschen mit normalen Genen, besteht praktisch kein Risiko. Wenn Ihr Partner das weniger schwer betroffene Gen trägt (eine Chance von 1 zu 10), beträgt das Risiko, dass ein Kind EPP erbt, eins zu vier. Wir empfehlen Ihnen, eine genetische Beratung bzw. die Konsultation eines Porphyriezentrums.
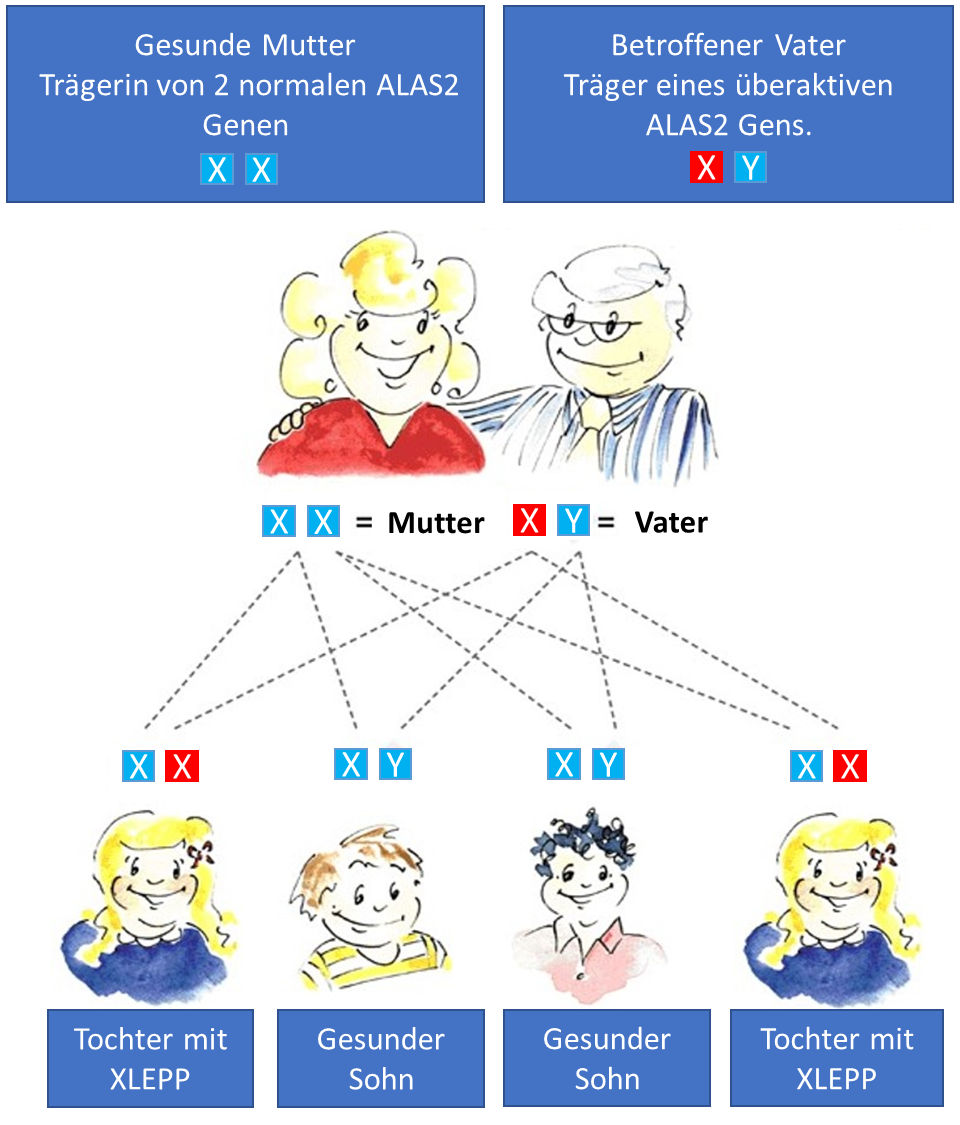
Das Gen für ALAS2, das bei XLP überaktiv ist, liegt auf dem X-Chromosom. Dieses X-Chromosom entscheidet zusammen mit einem zweiten X- oder einem Y-Chromosom über das Geschlecht eines Kindes (XX-Chromosomen sind weiblich, XY-Chromosomen sind männlich). Im Fall von XLP reicht ein übermäßig aktives Gen aus, um die Krankheit auszulösen. Väter mit XLP geben ihren Zustand an alle ihre Töchter weiter, können ihn aber nicht an einen Sohn weitergeben (der das nicht betroffene Y-Chromosom seines Vaters erbt). (siehe unten)
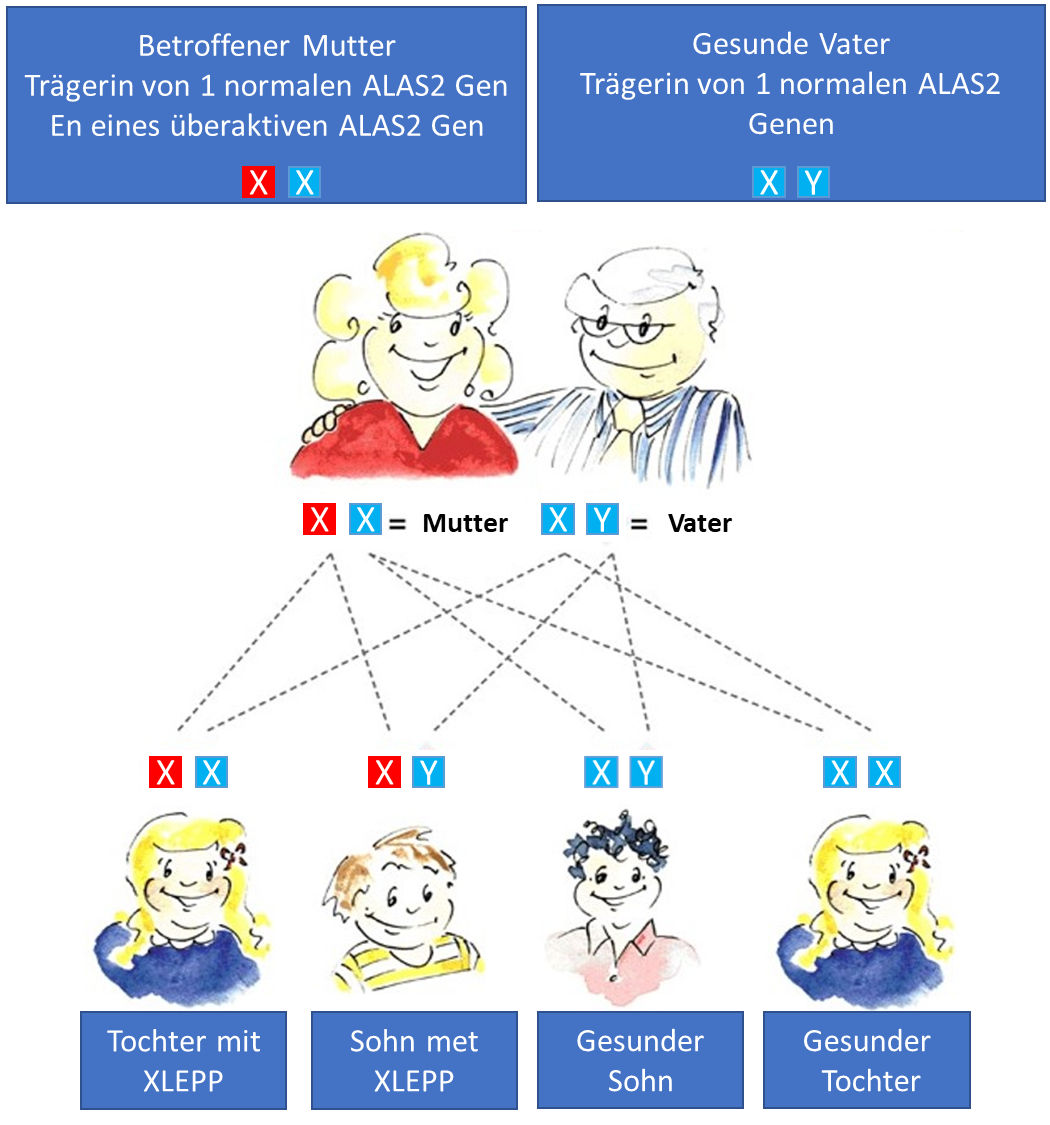
Mütter, die von XLP betroffen sind, geben ihr überaktives ALAS2-Gen an die Hälfte ihrer Töchter und Söhne weiter. Im Durchschnitt sind 50 % der Nachkommen eines betroffenen Elternteils von XLP betroffen. (siehe unten)
Sehr selten kann EPP oder XLP später im Leben auftreten, oft in Verbindung mit einer zugrunde liegenden bösartigen Bluterkrankung.
7. Sind EPP und XLP gefährliche Krankheiten?
Eine kleine Anzahl von EPP- und XLP-Patienten kann Leberschäden entwickeln. Es ist nicht vorhersagbar, wer davon betroffen sein wird, aber das ist zum Glück selten. Wenn Sie lichtempfindlicher werden, müde werden oder das Weiße in Ihren Augen gelb wird, sollten Sie ihre Ärztin oder ihren Arzt so schnell wie möglich um einen Leberfunktionstest bitten. EPP- und XLP-Patienten bekommen auch häufiger Gallensteine als die Allgemeinbevölkerung.Schwangerschaft und Geburt sind mit keinen zusätzlichen Risiken verbunden und für ein Baby kann eine normale Entwicklung erwartet werden. Manchmal können Frauen mit EPP während der Schwangerschaft länger im Sonnenlicht bleiben.
Da EPP-Patienten Sonnenlicht meiden, haben sie oft zu wenig Vitamin D und ihre Knochen enthalten daher weniger Kalzium als normal.
EPP-Patienten sind häufig leicht anämisch, mit niedrigen Hämoglobin- und Eisenwerten im Blut. Die Einnahme von Eisenpräparaten kann jedoch die Lichtempfindlichkeit erhöhen, und Eisenpräparate sollten nur auf ärztlichen Rat ein-genommen werden (siehe unten).
Sie sollten regelmäßig – mindestens einmal im Jahr - Ihren Arzt für Kontrolluntersuchungen aufsuchen. Ihre Ärztin bzw. ihr Arzt kann ihr Protoporphyrin im Blut, ihren Vitamin-D-Spiegel, ihre Leberfunktion und andere Organfunktionen durch jährliche Bluttests überwachen. Der Zweck der regelmäßigen Überwachung besteht darin, frühzeitig Veränderungen zu erkennen, bevor diese zu gesundheitlichen Risiken werden.
8. Lichtschutz in EPP und XLP
Von Kindheit an versuchen Patienten mit EPP, unnötige Sonneneinstrahlung und starke künstliche Lichtquellen zu vermeiden. Schutzkleidung wie Mützen, lange Ärmel, Handschuhe und Hosen sind von Vorteil. Dunkle und engmaschige Stoffe schützen besser als dünne Kleidung mit hellen Farben. Sonnenschutzmittel schützen hauptsächlich vor UV-Strahlung und nicht vor dem sichtbaren Licht, das die EPP-Symptome verursacht. Fenster in Häusern und Autos bieten keinen Schutz, da schädliches Licht durch die meisten Fenster dringt. Lehrkräfte in Schulen müssen über die Diagnose und die Notwendigkeit besonderer Anforderungen, wie ein fensterferner Arbeitsplatz und das Recht auf Vermeidung von Aktivitäten im Freien aufgeklärt werden.9. Wie können EPP und XLP behandelt werden?
Afamelanotid (Scenesse®) ist derzeit die einzige Therapie, für die Sicherheit und Wirksamkeit in klinischen Studien nachgewiesen wurde. Das Medikament wurde 2014 in der Europäischen Union und 2019 in den USA zugelassen. Die Behandlung mit Afamelanotid erhöht die Zeit, in der EPP-und XLP Patient:innen schmerzfrei im direkten Sonnenlicht verbringen können, und ihre Lebensqualität signifikant. Afamelanotid wird als Implantat verabreicht, das alle zwei Monate unter die Haut oberhalb der Hüfte injiziert wird. Es wurde von leichten Nebenwirkungen berichtet. Afamelanotid ist noch nicht in allen europäischen Ländern erhältlich.Substanzen und Interventionen mit derzeit nicht nachgewiesener Wirksamkeit
Eine Vielzahl von Interventionen und Substanzen wurde verwendet, um die Symptome von EPP und XLP zu verbessern, obwohl Sicherheit und Wirksamkeit nicht durch klinische Studien untersucht wurden. Im Folgenden werden die am häufigsten verwendeten Substanzen und Interventionen diskutiert.
• Beta-Carotin
Beta-Carotin wurde früher für EPP verschrieben, aber nur wenige Menschen mit EPP fanden die Einnahme hilfreich. Beta-Carotin wird aus einer Substanz gewonnen, die Karotten orange färbt und der Haut normalerweise einen leicht orangefarbenen Ton verleiht. Sie sollten die Risiken und Vorteile dieser Option mit Ärzt:innen in einem Porphyrie-Zentrum besprechen, bevor Sie mit der Behandlung beginnen.
• Antihistaminika
Antihistaminika, die vor allem bei Allergien eingesetzt werden, können bei einem Nesselausschlag helfen. Einige Patienten berichten von einer zusätzlichen positiven Wirkung von Hydrocortison-Creme auf geröteter oder geschwollener Haut. Ältere Antihistaminika, bei denen Müdigkeit eine Nebenwirkung ist, können nach einer leichten phototoxischen Reaktion auch beim Schlafen helfen.
• Phototherapie
Die Schmalband-UV-B- und PUV-A-Therapie sind Behandlungen mit ultraviolettem Licht, die in dermatologischen Abteilungen bei verschiedenen Hauterkrankungen durchgeführt werden. Bei EPP wurden sie verwendet, um die Lichttoleranz zu erhöhen, indem sie die Haut leicht verdicken und eine Bräune entwickeln. Die Therapie beinhaltet die sorgfältige Exposition gegenüber steigenden Dosen kontrollierter UV-B-Strahlung, normalerweise dreimal pro Woche für mindestens fünf Wochen im Frühjahr. Dies kann für einige Patient:innen unpraktisch und unbequem sein. Die Phototherapie sollte immer von ausgebildeten Photodermatolog:innen durchgeführt werden. Es wird dringend davon abgeraten, Selbstbräuner zu benutzen, da diese photo-toxische Reaktionen auslösen können.
• Andere Behandlungen werden derzeit geprüft.
10. Unterstützende Therapien und Interventionen
• Linderung einer schmerzhaften phototoxischen ReaktionDer starke brennende Schmerz in der Haut nach Lichteinwirkung ist schwer zu lindern. Viele EPP-Patienten berichten von einer vorübergehenden Linderung durch kalte Bäder, kalte nasse Verbände oder Heilmittel wie Aloe Vera. Einige Patienten berichten von einer erheblichen Schmerzlinderung durch warmes Wasser oder Wasserdampf. Schmerzmittel sind nicht besonders wirksam. Die Auswahl und Austestung von Schmerzmitteln sollte gemeinsam mit ihrer Ärztin oder ihrem Arzt erfolgen.
• Vitamin-D
Vitamin D wird hauptsächlich in sonnenexponierter Haut produziert, aber auch in Lebensmitteln wie fettem Fisch, Eiern, Fleisch und Getreide. Personen, die die Sonne meiden, entwickeln häufig einen Vitamin-D-Mangel und alle EPP-Patienten sollten Vitamin D in der empfohlenen Dosis einnehmen.
• Eisenpräparate
Viele Patienten mit EPP haben leicht niedrige Eisen- (Ferritin-) und Hämoglobinwerte. Es kann daher schwierig sein herauszufinden, ob Sie eine echte Eisenmangelanämie haben und eine Eisenergänzung benötigen. Der Vergleich mit früheren Blut- und Eisenwerten hilft.
Ein Eisenmangel, der eine Anämie verursacht, sollte wie bei jedem anderen Patienten beachtet werden. Bei EPP kann eine Eisenergänzung jedoch die Produktion von roten Blutkörperchen im Knochenmark beschleunigen und den Protoporphyrinspiegel im Blut erhöhen. Dies kann zu einer erhöhten Lichtempfindlichkeit führen. Dies ist bei XLP nicht der Fall, da Eisenpräparate hier zu einer Senkung des Protoporphyrinspiegels im Blut führen.
• Sonnenschutzmittel
Da sichtbares Licht die Probleme bei EPP und XLP verursacht, sind gängige Sonnenschutzmittel, die vor ultraviolettem Licht (insbesondere UV-B) schützen, nicht sehr wirksam.
• Lichtfilter
Die meisten Fensterfilter für Autos und Büros sind so konzipiert, dass sie ultraviolettes Licht blockieren. Auch wenn sie in rauchigen oder grauen Farben getönt sind, lassen sie den Großteil des sichtbaren Lichts durch. Filter, die selektiv das schädlichste Licht für die EPP blockieren, sind gelb. Diese Filter (Folien) können auf Fenster in Wohnungen, Schulen oder Büros oder sogar auf Seitenfenstern von Autos aufgebracht werden. Bevor Sie solche Folien auf Autoscheiben anbringen, sollten Sie sich bei den Verkehrsbehörden nach den entsprechenden Verkehrsvorschriften erkundigen.
• Leberschutz
Übermäßiger Konsum von Alkohol oder anderen leberschädigenden Substanzen ist zu vermeiden und sie sollten sich gegen Hepatitis A und B impfen lassen.
11. Zusätzliche Informationen
• Können bestimmte Medikamente EPP verschlimmern?EPP unterscheidet sich von den akuten hepatischen Porphyrien, die durch bestimmte Medikamente verschlimmert werden können. Sofern keine Allergie gegen ein Medikament vorliegt, können Personen mit EPP alle Medikamente einnehmen, die ihre Gesundheit erfordert.
• Laserbehandlung
Laserbehandlungen für Augenoperationen oder Haarentfernung wurden nicht als Problem bei EPP beschrieben. Die blauen Laser, die bei der Reparatur von Zahnhöhlen verwendet werden, können Verbrennungen im Zahnfleisch verursachen. Zahnärzte sollten über die EPP-Diagnose informiert werden, damit sie den Laserstrahl vorsichtig einstellen können.
• Lichtquellen im Gesundheitswesen
Das Abdecken der Operationsleuchten mit Gelbfiltern ist bei den meisten Operationen nicht erforderlich, jedoch sollte dem Pflegepersonal immer mitgeteilt werden, dass Sie eine EPP-Diagnose haben, um unnötig starke Beleuchtung bei längeren Operationen zu vermeiden. Auch die Untersuchung des Körperinneren mit einem beleuchteten, flexiblen Instrument, einem sogenannten Endoskop, ist sicher.
Vor Operationen oder Endoskopien bei Patient:innen mit schwerer EPP-bedingter Lebererkrankung, sollten sich die Ärzt:innen mit einem Porphyriezentrum beraten, da die Operationslampen mit Gelbfiltern abgedeckt werden sollten. Porphyriezentren bieten bei Bedarf weiterführende Beratung an, falls notwendig.
12. Können EPP und XLP geheilt werden?
Gegenwärtig gibt es keine andere Heilung für EPP als die Knochenmarktransplantation. Diese Behandlung ist mit einem so hohen Risiko mit Komplikationen bis hin zum Tod verbunden, dass sie nur in sehr speziellen und komplizierten Situationen zum Einsatz kommt.13. Wo erhalte ich weitere Informationen zu EPP und XLP?
Die Informationen auf dieser Website wurden von einer Gruppe von Porphyrie-Spezialisten verfasst und aktualisiert, die in Ipnet zusammenarbeiten und von Vertretern von Patientenorganisationen geprüft wurden.Obwohl im Internet viele andere Informationsquellen zu finden sind, wurden diese möglicherweise nicht von Porphyrie-Spezialisten verfasst oder auf Richtigkeit hin geprüft.
Selbsthilfegruppen können eine gute Anlaufstelle für Informationen, Vernetzung und Unterstützung sein. Porphyrie-Patientengruppen in verschiedenen europäischen Ländern sind auf der Website des International Porphyria Network und der Website der Global Porphyria Advocacy Coalition (GPAC) aufgeführt.
Überarbeitet: Mai 2021
Englisch-Deutsch-Übersetzung:
Prof. Dr. med. Ulrich Stölzel (Porphyriezentrum Chemnitz), Dr. Sebastian Reuber (Berliner Leberring) November 2021